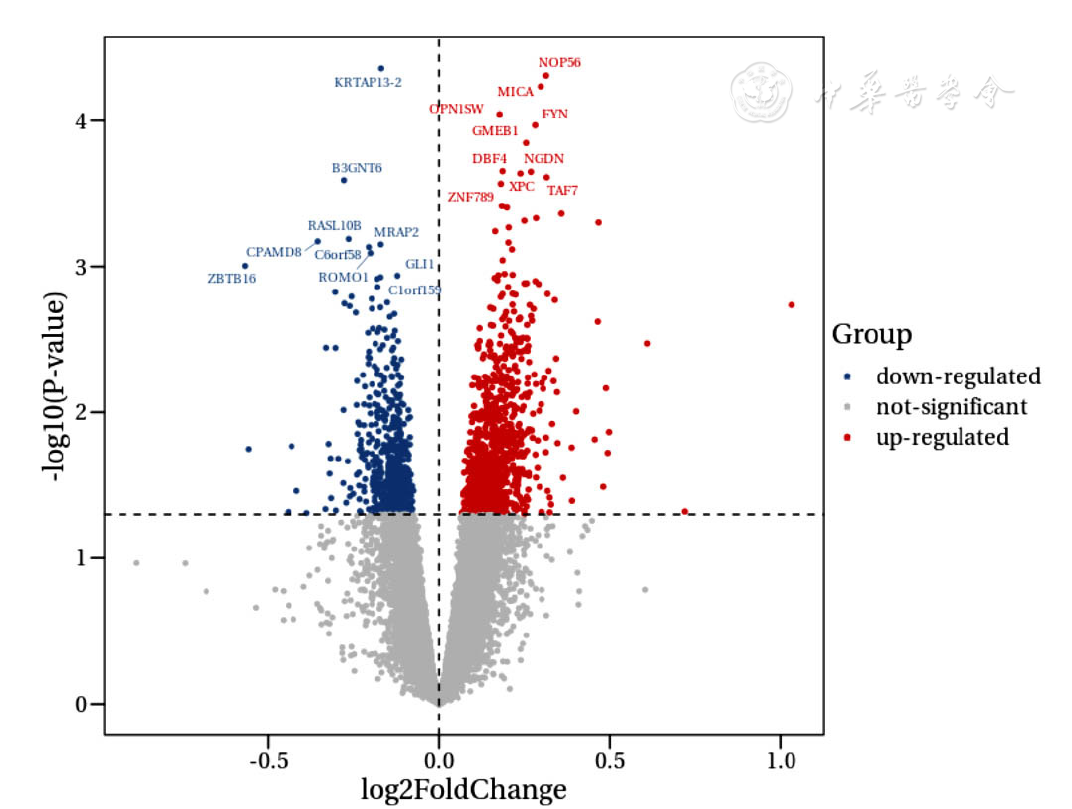
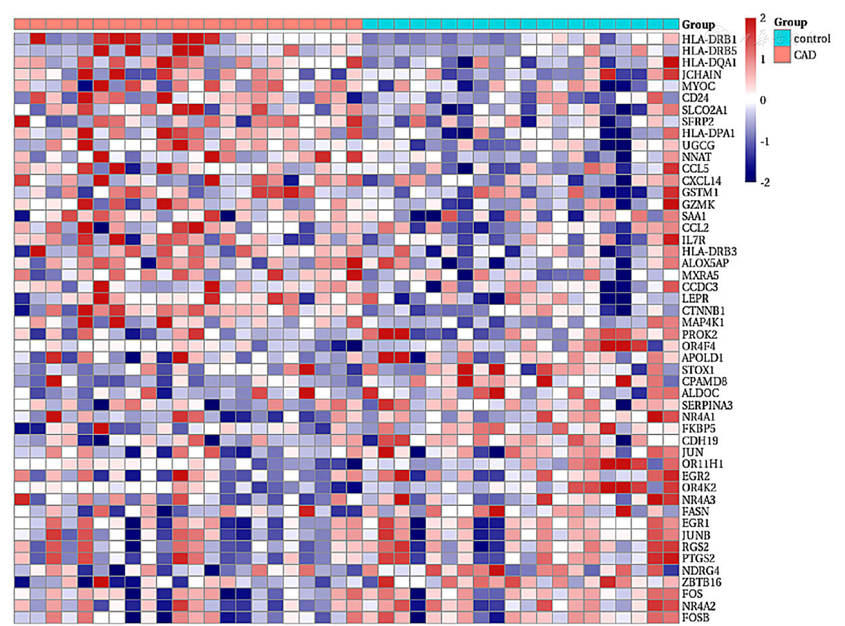
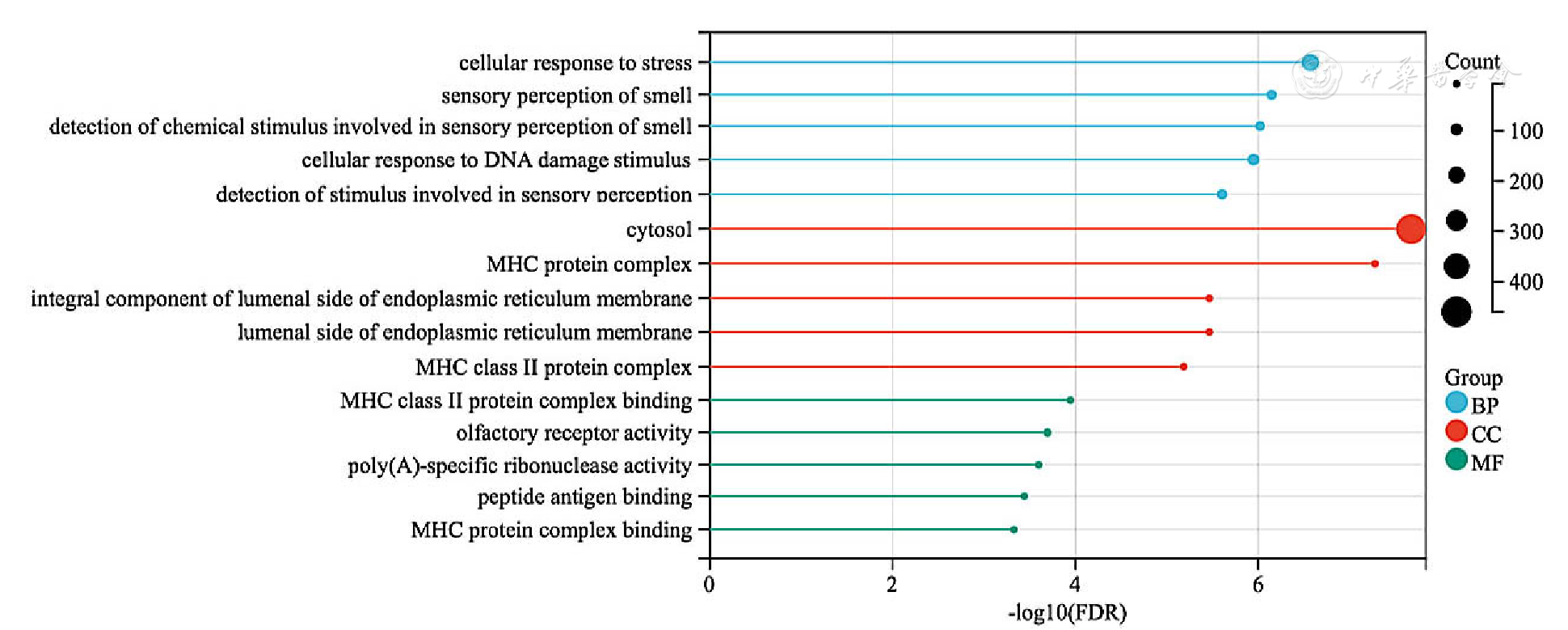
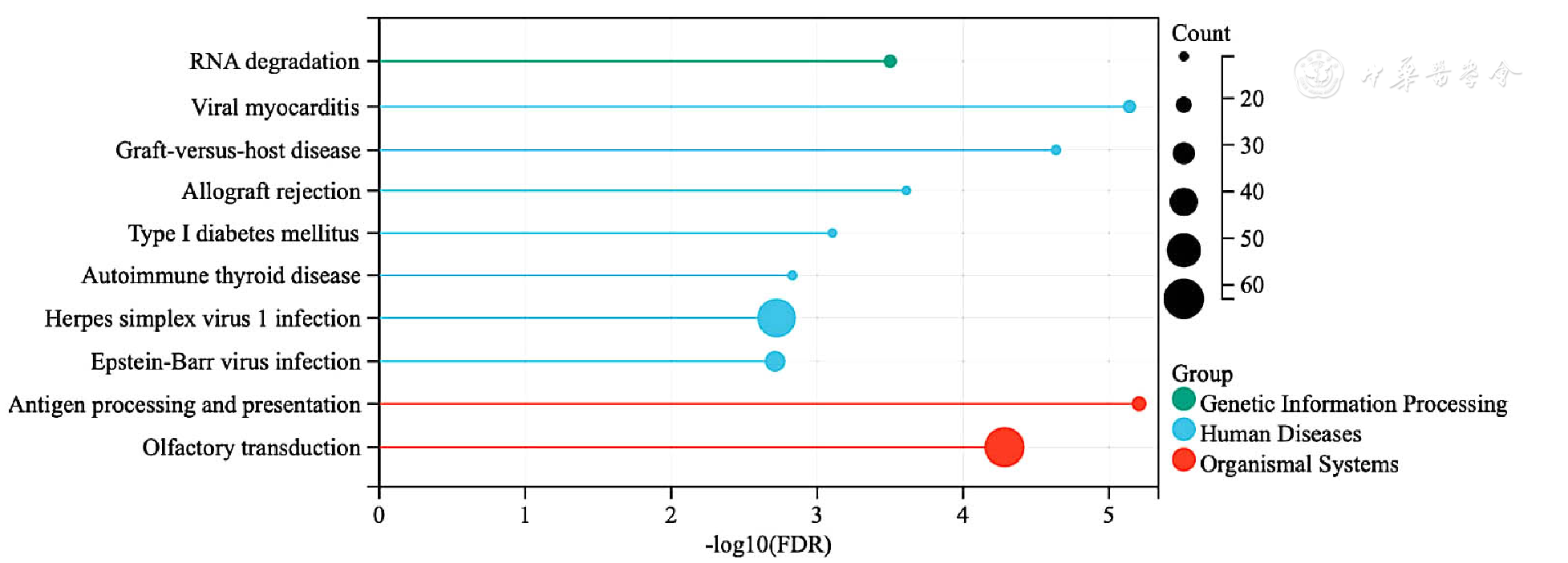
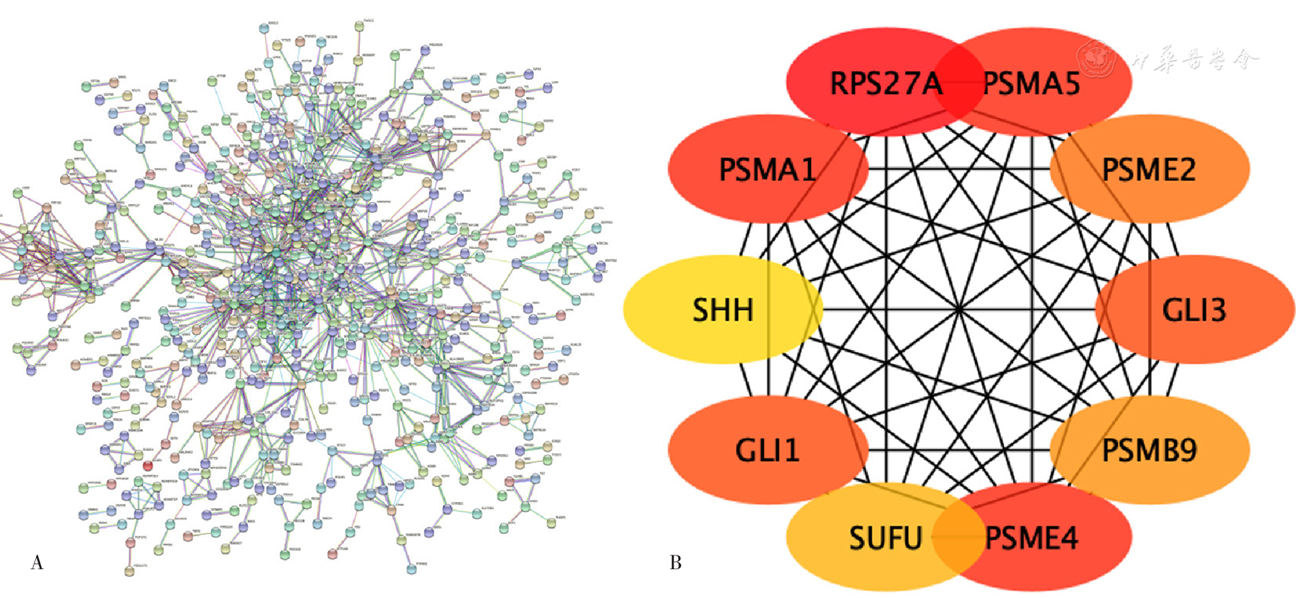
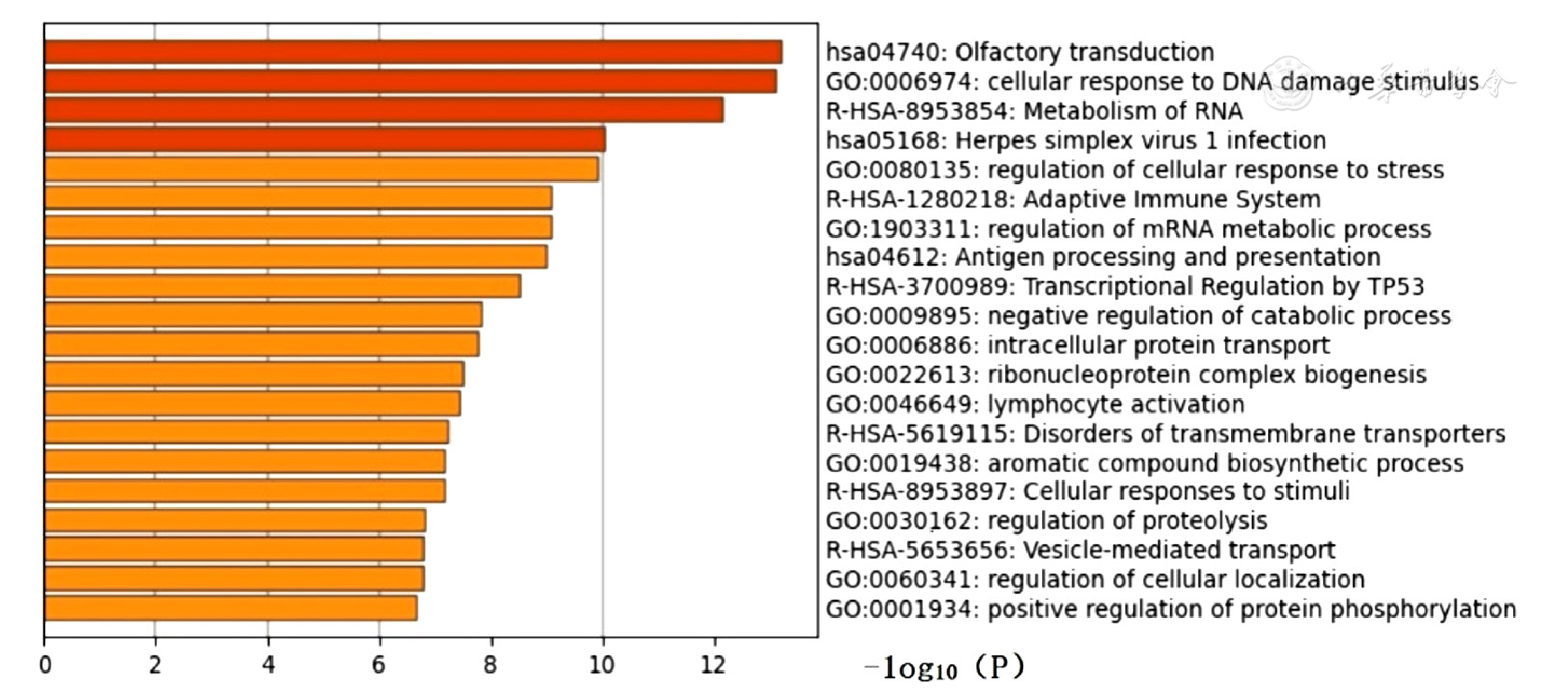
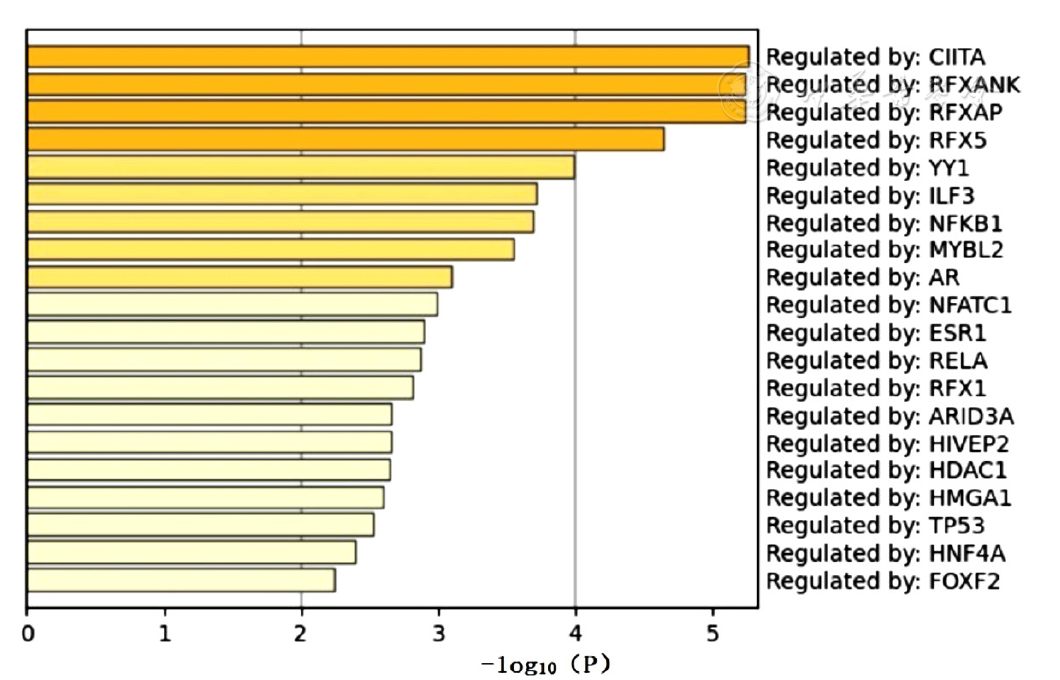
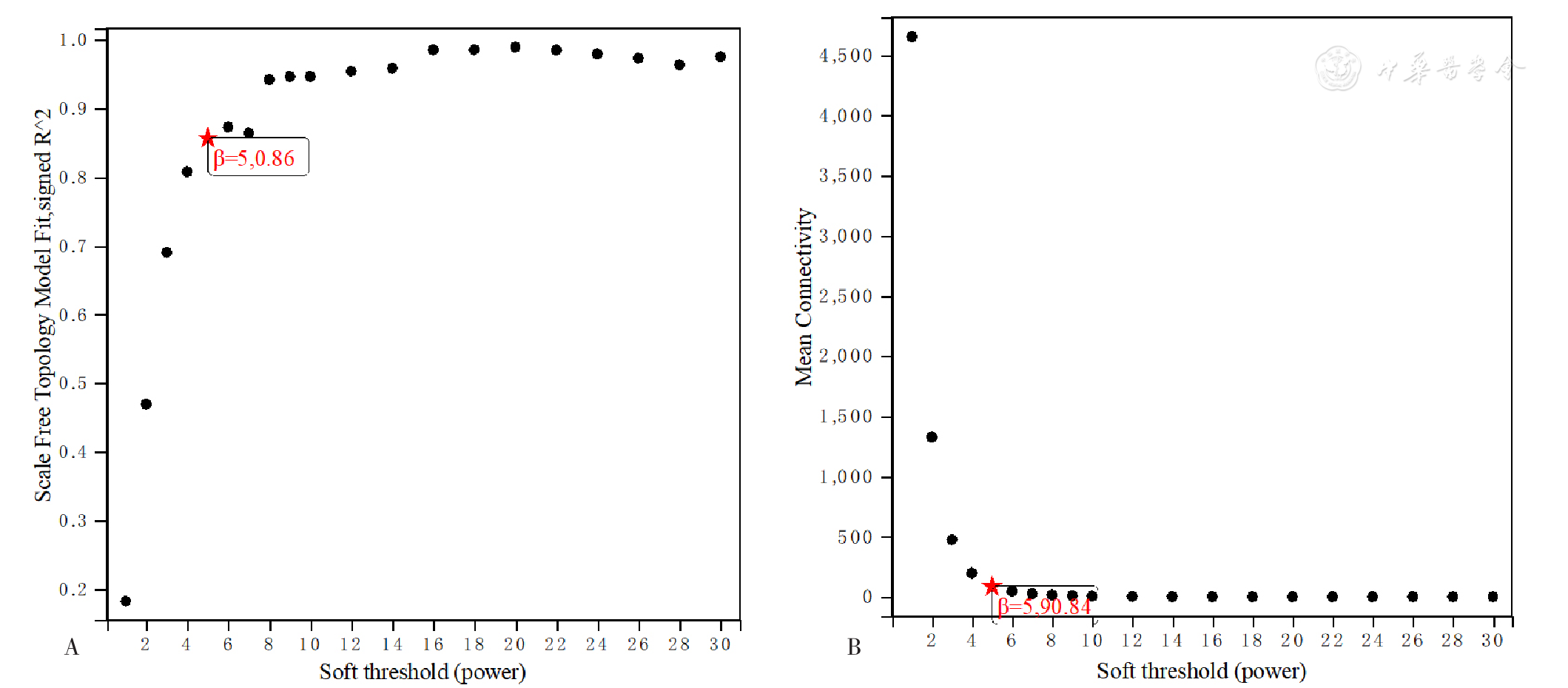
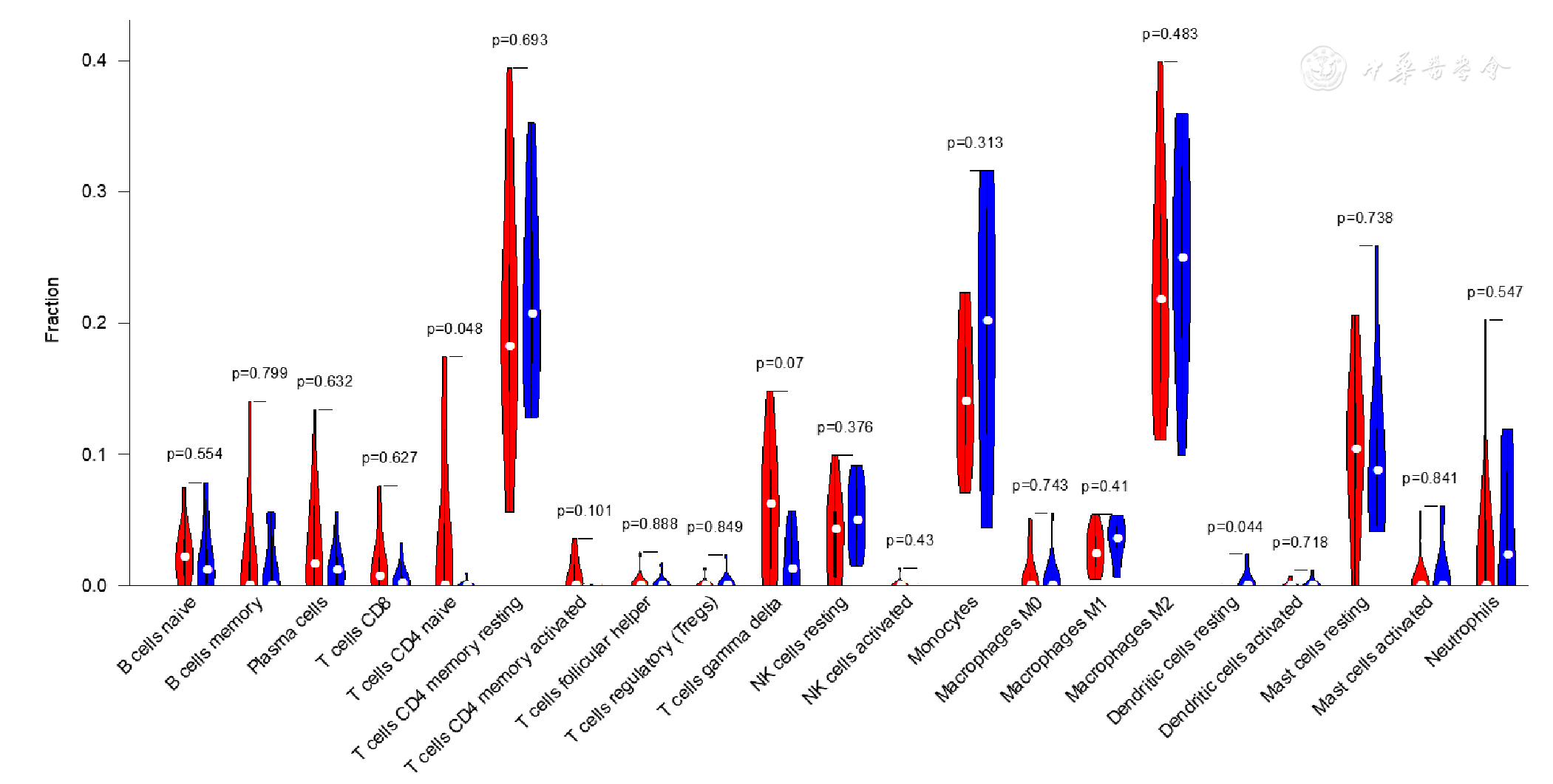
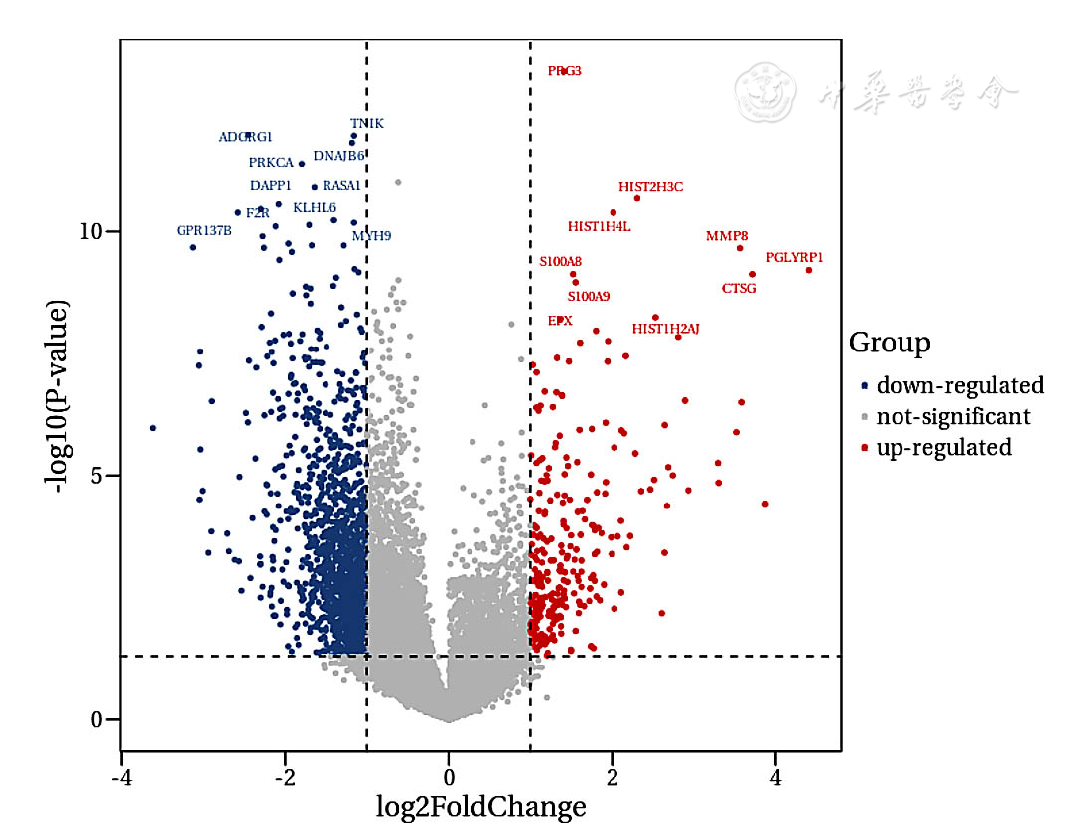
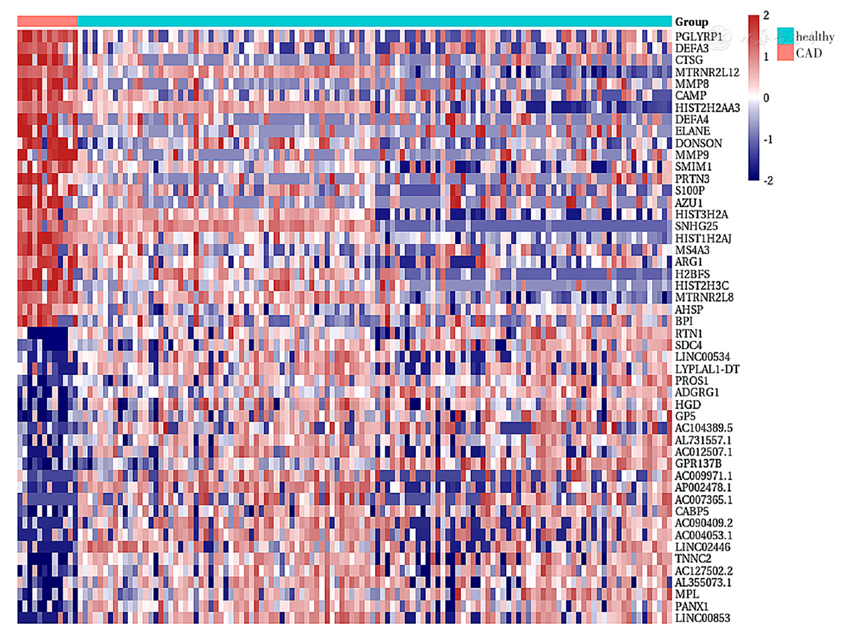
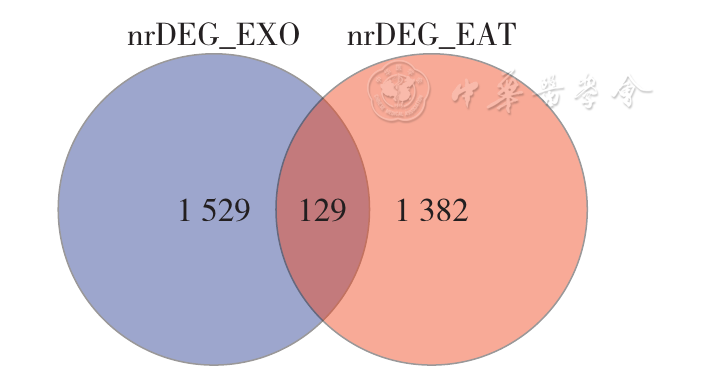
| [1] |
NAPOLI C, CRUDELE V, SORICELLI A,et al. Primary prevention of atherosclerosis:a clinical challenge for the reversal of epigenetic mechanisms? [J]. Circulation, 2012, 125(19):2363-2373. DOI: 10.1161/CIRCULATIONAHA.111.085787.
|
| [2] |
ALKHALIL M, CHOUDHURY R P. Current concepts in atherosclerosis[J]. Indian J Thorac Cardiovasc Surg, 2018, 34(Suppl 3):198-205. DOI: 10.1007/s12055-018-0699-y.
|
| [3] |
BEL J S, TAI T C, KHAPER N,et al. Mirabegron:the most promising adipose tissue beiging agent[J]. Physiol Rep, 2021, 9(5):e14779. DOI: 10.14814/phy2.14779.
|
| [4] |
PATEL K H K, HWANG T, SE LIEBERS C,et al. Epicardial adipose tissue as a mediator of cardiac arrhythmias[J]. Am J Physiol Heart Circ Physiol, 2022, 322(2):H129-144. DOI: 10.1152/ajpheart.00565.2021.
|
| [5] |
CONTE M, PETRAGLIA L, POGGIO P,et al. Inflammation and cardiovascular diseases in the elderly:the role of epicardial adipose tissue[J]. Front Med (Lausanne), 2022, 9:844266. DOI: 10.3389/fmed.2022.844266.
|
| [6] |
SONG Y, SONG F, WU C,et al. The roles of epicardial adipose tissue in heart failure[J]. Heart Fail Rev, 2022, 27(1):369-377. DOI: 10.1007/s10741-020-09997-x.
|
| [7] |
ZHAO Q W, YANG J, LIU B L,et al. Exosomes derived from mangiferinstimulated perivascular adipose tissue ameliorate endothelial dysfunction[J]. Mol Med Rep, 2019, 19(6):4797-4805. DOI: 10.3892/mmr.2019.10127.
|
| [8] |
SMYTH G K, MICHAUD J, SCOTT H S. Use of within-array replicate spots for assessing differential expression in microarray experiments[J]. Bioinformatics, 2005, 21(9):2067-2075. DOI: 10.1093/bioinformatics/bti270.
|
| [9] |
YU G, WANG L G, HAN Y,et al. clusterProfiler:an R package for comparing biological themes among gene clusters[J]. OMICS, 2012, 16(5):284-287. DOI: 10.1089/omi.2011.0118.
|
| [10] |
ZHOU Y Y, ZHOU B, PACHE L,et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets[J]. Nat Commun, 2019, 10(1):1523. DOI: 10.1038/s41467-019-09234-6.
|
| [11] |
SHANNON P, MARKIEL A, OZIER O,et al. Cytoscape:a software environment for integrated models of biomolecular interaction networks[J]. Genome Res, 2003, 13(11):2498-2504. DOI: 10.1101/gr.1239303.
|
| [12] |
OIKONOMOU E K, WILLIAMS M C, KOTANIDIS C P,et al. A novel machine learning-derived radiotranscriptomic signature of perivascular fat improves cardiac risk prediction using coronary CT angiography[J]. Eur Heart J, 2019, 40(43):3529-3543. DOI: 10.1093/eurheartj/ehz592.
|
| [13] |
SATO T, AIZAWA Y, YUASA S,et al. The effect of dapagliflozin treatment on epicardial adipose tissue volume[J]. Cardiovasc Diabetol, 2018, 17(1):6. DOI: 10.1186/s12933-017-0658-8.
|
| [14] |
SAGE A P, MURPHY D, MAFFIA P,et al. MHC Class II-restricted antigen presentation by plasmacytoid dendritic cells drives proatherogenic T cell immunity[J]. Circulation, 2014, 130(16):1363-1373. DOI: 10.1161/CIRCULATIONAHA.114.011090.
|
| [15] |
FAN Z W, LI J F, LI P,et al. Protein arginine methyltransferase 1 (PRMT1)represses MHCⅡtranscription in macrophages by methylating CIITA[J]. Sci Rep, 2017, 7:40531. DOI: 10.1038/srep40531.
|
| [16] |
WU X Y, KONG X C, CHEN D W,et al. SIRT1 links CIITA deacetylation to MHCⅡ activation[J]. Nucleic Acids Res, 2011, 39(22):9549-9558. DOI: 10.1093/nar/gkr651.
|
| [17] |
KONG X C, FANG M M, LI P,et al. HDAC2 deacetylates class Ⅱ transactivator and suppresses its activity in macrophages and smooth muscle cells[J]. J Mol Cell Cardiol, 2009, 46(3):292-299. DOI: 10.1016/j.yjmcc.2008.10.023.
|
| [18] |
IBORRA S, GONZÁLEZ-GRANADO J M. In vitro differentiation of naïve CD 4+ T cells:a tool for understanding the development of atherosclerosis[J]. Methods Mol Biol, 2015, 1339:177-189. DOI: 10.1007/978-1-4939-2929-0_12.
|
| [19] |
GADDIS D E, PADGETT L E, WU R P,et al. Atherosclerosis impairs naive CD 4+ T-cell responses via disruption of glycolysis[J]. Arterioscler Thromb Vasc Biol, 2021, 41(9):2387-2398. DOI: 10.1161/ATVBAHA.120.314189.
|
| [20] |
XIA X D, WANG M M, LI J,et al. Identification of potential genes associated with immune cell infiltration in atherosclerosis[J]. Math Biosci Eng, 2021, 18(3):2230-2242. DOI: 10.3934/mbe.2021112.
|
| [21] |
HUANG J, ZHOU Q. Identification of the relationship between hub genes and immune cell infiltration in vascular endothelial cells of proliferative diabetic retinopathy using bioinformatics methods[J]. Dis Markers, 2022, 2022:7231046. DOI: 10.1155/2022/7231046.
|
| [22] |
LI Y N, GUO X J, XUE G H,et al. RNA Splicing of the Abi1 Gene by MBNL1 contributes to macrophage-like phenotype modulation of vascular smooth muscle cell during atherogenesis[J]. Cell Prolif, 2021, 54(5):e13023. DOI: 10.1111/cpr.13023.
|
| [23] |
CHEN A Q, GAO X F, WANG Z M,et al. Therapeutic exosomes in prognosis and developments of coronary artery disease[J]. Front Cardiovasc Med, 2021, 8:691548. DOI: 10.3389/fcvm.2021.691548.
|
| [24] |
BOND S T, CALKIN A C, DREW B G. Adipose-derived extracellular vesicles:systemic messengers and metabolic regulators in health and disease[J]. Front Physiol, 2022, 13:837001. DOI: 10.3389/fphys.2022.837001.
|
| [25] |
AHMADIEH S, KIM H W, WEINTRAUB N L. Potential role of perivascular adipose tissue in modulating atherosclerosis[J]. Clin Sci (Lond), 2020, 134(1):3-13. DOI: 10.1042/CS20190577.
|
| [26] |
LIU Y, SUN Y, LIN X Z,et al. Perivascular adipose-derived exosomes reduce macrophage foam cell formation through miR-382-5p and the BMP4-PPARγ-ABCA1/ABCG1 pathways[J]. Vascul Pharmacol, 2022, 143:106968. DOI: 10.1016/j.vph.2022.106968.
|
| [27] |
LI X Z, BALLANTYNE L L, YU Y,et al. Perivascular adipose tissue-derived extracellular vesicle miR-221-3p mediates vascular remodeling[J]. FASEB J, 2019, 33(11):12704-12722. DOI: 10.1096/fj.201901548R.
|
| [28] |
MARKOVICD, JEVTOVIC-STOIMENOV T, COSIC V,et al. Clinical utility of survivin (BIRC5),novel cardiac biomarker,as a prognostic tool compared to high-sensitivity C-reactive protein,heart-type fatty acid binding protein and revised lee score in elderly patients scheduled for major non-cardiac surgery:a prospective pilot study[J]. J Med Biochem, 2018, 37(2):110-120. DOI: 10.1515/jomb-2017-0046.
|
| [29] |
BEDENBENDER K, SCHMECK B T. Endothelial ribonuclease 1 in cardiovascular and systemic inflammation[J]. Front Cell Dev Biol, 2020, 8:576491. DOI: 10.3389/fcell.2020.576491.
|
| [30] |
TAVAKOLIAN FERDOUSIE V, MOHAMMADI M, HASSANSHAHI G,et al. Serum CXCL10 and CXCL12 chemokine levels are associated with the severity of coronary artery disease and coronary artery occlusion[J]. Int J Cardiol, 2017, 233:23-28. DOI: 10.1016/j.ijcard.2017.02.011.
|
| [31] |
MAUSE S F, RITZEL E, DECK A,et al. Engagement of the CXCL12-CXCR4 axis in the interaction of endothelial progenitor cell and smooth muscle cell to promote phenotype control and guard vascular homeostasis[J]. Int J Mol Sci, 2022, 23(2):867. DOI: 10.3390/ijms23020867.
|
| [32] |
SNARSKI P, SUKHANOV S, YOSHIDA T,et al. Macrophage-specific IGF-1 overexpression reduces CXCL12 chemokine levels and suppresses atherosclerotic burden in apoe-deficient mice[J]. Arterioscler Thromb Vasc Biol, 2022, 42(2):113-126. DOI: 10.1161/ATVBAHA.121.316090.
|
| [33] |
LI F C, YAN K M, WU L L,et al. Single-cell RNA-seq reveals cellular heterogeneity of mouse carotid artery under disturbed flow[J]. Cell Death Discov, 2021, 7(1):180. DOI: 10.1038/s41420-021-00567-0.
|
| [34] |
HUBACEK J A, PITHA J, SKODOVÁ Z,et al. Polymorphisms in the lipopolysaccharide-binding protein and bactericidal/permeability-increasing protein in patients with myocardial infarction[J]. Clin Chem Lab Med, 2002, 40(11):1097-1100. DOI: 10.1515/CCLM.2002.191.
|
| [35] |
BLEIJERVELD O B, WIJTEN P, CAPPADONA S,et al. Deep proteome profiling of circulating granulocytes reveals bactericidal/permeability-increasing protein as a biomarker for severe atherosclerotic coronary stenosis[J]. J Proteome Res, 2012, 11(11):5235-5244. DOI: 10.1021/pr3004375.
|
| [36] |
COLE J W. F2R polymorphisms and clopidogrel efficacy and safety:another step toward precision medicine[J]. Neurology, 2021, 96(1):10-11. DOI: 10.1212/WNL.0000000000011085.
|

 )
)